



Qu’est-ce qu’un essai clinique ?
Les essais cliniques sont des études scientifiques effectuées chez l’homme en vue du développement des connaissances biologiques et médicales. En cancérologie, les essais cliniques impliquent les personnes atteintes d’un cancer.
Les essais cliniques sont aussi appelés « essais thérapeutiques » et font partie des « études cliniques ».
Ils définissent une situation expérimentale qui teste chez les patients de nouveaux médicaments, de nouvelles associations de médicaments, de nouveaux diagnostics (ex : test biologique), de nouvelles techniques de chirurgie, de radiothérapie, des dispositifs médicaux, de nouvelles façons d’administrer un traitement (ex : comprimé plutôt que perfusion), etc…
L’objectif des essais cliniques est de répondre à des questions non encore résolues ou de valider des hypothèses portant sur la tolérance, l’utilisation ou l’efficacité :
- Quelle est la dose du médicament pouvant être administrée sans effet indésirable important ?
- Le médicament /dispositif/stratégie thérapeutique (ou l’association de médicaments) est-il efficace ?
- Quels en sont les effets secondaires (toxicité) ?
Un médicament en cours de développement est appelé médicament expérimental.
Ces essais cliniques sont conduits par un promoteur (laboratoire pharmaceutique, centre hospitalier (Centre de Lutte contre le Cancer, Centre Hospitalier Universitaire) qui porte la responsabilité de l’étude.
Ils sont mis en œuvre par un médecin (appelé médecin investigateur) en collaboration avec une équipe médicale et de recherche, et dans le respect d’un cadre réglementaire éthique (protection et bien être des participants) et scientifique (méthodologie précise, bonnes pratiques, balance bénéfices/risques).
inclus dans un essai clinique au CLB en 2025
soit 19,1% de la file active
Essais Cliniques : un outil pour vous accompagner
Recherchez les essais cliniques ouverts au Centre Léon Bérard

Comment se déroulent les étapes du développement d’un médicament ?
Le développement d'un médicament contre le cancer (chimiothérapie, immunothérapie, thérapie ciblée...) est un parcours long.
Afin d’obtenir une autorisation pour être prescrit aux patients (Autorisation de Mise sur le Marché ou AMM) un médicament suit un circuit de développement qui comprend plusieurs étapes, de la phase préclinique aux phases cliniques.
Une quinzaine d’années s’écoulent entre les premiers travaux scientifiques (recherche fondamentale) et la commercialisation d’un médicalement. Un parcours long qui aura raison de nombreuses molécules se révélant trop toxiques et/ou inefficaces.
Environ 1 seule molécule sur 1000 sera finalement mise sur le marché. Un médicament peut être disponible en France mais pas dans un autre pays, ou vice-versa. En effet, le dossier d’AMM d’un médicament doit être validé par les autorités de santé spécifiques (Agence Européenne du Médicament en Europe, Agence Nationale de Sécurité du Médicament en France) d’un continent et d’un pays.
A noter qu’en France, pour être remboursé par la Sécurité Sociale, un médicament doit également obtenir un avis favorable de commissions spécialisées (Commission de la Transparence et Commission d’évaluation économique et de santé publique de l’HAS *).
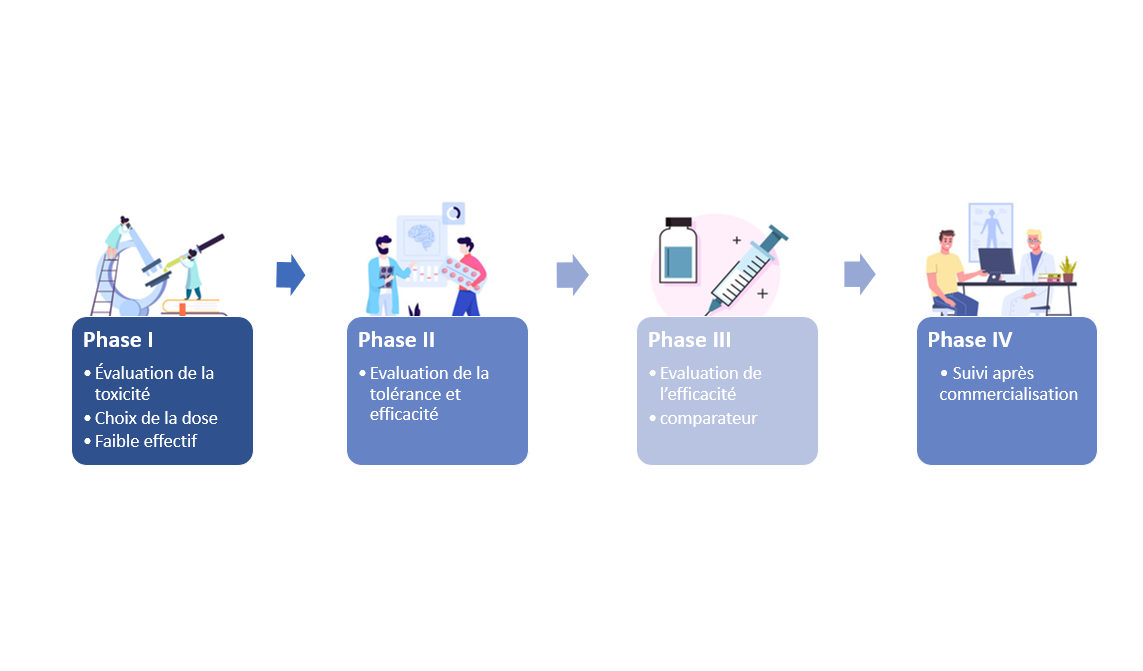
Phase préclinique
Il s’agit, dans le cas de médicaments, de recherches faites en laboratoire sur des cellules (recherche in vitro) et chez l’animal (recherche in vivo). Cette phase permet de connaître le mécanisme d’action de la molécule et ses potentiels effets toxiques (fertilité, effet mutagène…) et d’étudier son devenir de son absorption à son élimination. Des résultats dans cette phase sont nécessaires pour autoriser les premiers essais cliniques chez l’homme.
Phase I « évaluation de la toxicité/choix de la dose »
Les essais cliniques de phase I sont effectués dans certaines structures habilitées par les Agences Régionales de Santé (ARS). C’est le cas du Centre Léon Bérard qui est également un centre labellisé par l’Institut National du Cancer (INCa) pour mener ces essais en cancérologie. L’autorisation de lieu de recherche garantit que ces études sont prises en charge par une équipe médicale spécialisée dans un environnement spécialement organisé pour prendre en charge ce type d’essais cliniques complexes.
Ces essais évaluent les premières administrations du médicament ou d’une association de médicaments chez l’homme. Ils permettent de déterminer la dose optimale à administrer, étudient les manifestations qui surviennent chez les participants (toxicités éventuelles) et de quelle façon se comporte le médicament dans l’organisme de son absorption à son élimination.
Ces essais cliniques appelés précoces impliquent peu de patients (en général entre 20 et 80) mais pouvant présenter en cancérologie des tumeurs de localisations différentes. Ils sont complexes à mettre en place et ont de nombreuses procédures à suivre (prises de sang, électrocardiogrammes, biopsies de la tumeur, visites médicales,…).
-
 15 Avr
15 AvrCancer du rein avec métastases cérébrales : des résultats prometteurs avec le cabozantinib
Une étude récente publiée dans le journal scientifique European Journal of Cancer met en lumière des résultats encourageants pour les patients atteints de cancer du rein avec métastases cérébrales, une situation clinique particulièrement complexe à traiter.
Lire l'article -

Essais cliniques de phase 1 : 3 questions au Dr Cassier
Les essais cliniques de phase 1, expliqué par le Dr Cassier.
Lire l'article
Phase II « évaluation de la tolérance et de l’efficacité »
Ces essais concernent un plus grand nombre de patients, présentant en cancérologie un type homogène de tumeur. Ils vont permettre de confirmer la dose optimale du médicament (ou de l’association de médicaments) déterminée lors des phases I, d’améliorer la compréhension des effets secondaires et d’obtenir des données d’efficacité préliminaires.
Phase III « essais comparatifs »
Ces essais concernent plusieurs centaines jusqu’à plusieurs milliers de participants selon les situations. Ils servent à démontrer l’intérêt du traitement et à comparer son efficacité par rapport soit à un placebo, soit au traitement de référence (traitement déjà prouvé efficace et prescrit dans la même indication).
Ces essais comparatifs utilisent le tirage au sort des médicaments à l’étude, afin qu’un patient ait la même probabilité de recevoir le médicament de référence (ou le placebo) que le médicament expérimental. Ce tirage au sort est appelé « randomisation » et permet de rendre homogène les groupes de patients qui seront comparés pour évaluer l’efficacité du médicament expérimental par rapport au médicament de référence.
De cette façon, si une différence d’efficacité est constatée à la fin de l’essai en faveur du médicament expérimental, elle ne pourra être attribuée qu’à une activité biologique supérieure de ce médicament, et non à des différences entre les caractéristiques des patients.
Phase IV « évaluation après commercialisation »
L’évaluation des médicaments ne s’arrête pas avec les résultats des essais de phase III et l’AMM. En effet, la tolérance et l’activité des médicaments en pratique courante, c’est-à-dire sur la population d’usage des traitements. Cette population d’usage est beaucoup plus diversifiée que celle des essais cliniques (par exemple : présence d’autres maladies chroniques, prise simultanée d’autres traitements…). C’est pour cela que la survenue d’éventuels effets secondaires très rares, inattendus ou à distance, est enregistrée au niveau national dans le cadre du réseau de pharmacovigilance ; et que des études cliniques dites « en vie réelle » permettent de document l’efficacité des traitements commercialisés en conditions réelles d’utilisation.
Quand, pourquoi et comment participer à un essais clinique ?
Tout au long de votre parcours médical, votre médecin référent ou un autre intervenant (radiologie, service des urgences imprévues, soins de support) peut vous proposer de participer à un essai clinique.
Ces essais peuvent se dérouler à toutes les phases de votre parcours : avant ou au cours de la chirurgie, lors des traitements de chimiothérapie, de radiothérapie, et même pendant la phase de surveillance. Chaque essai est proposé afin de répondre à la question spécifique posée dans l’essai clinique.
Il concerne une population bien définie avec des critères d’entrée dans l’essai clinique que l’on appelle critères d’éligibilité. Ces critères sont propres à chaque essai clinique, ce qui explique que l’essai ne peut pas être proposé à tous les patients.
Ces critères sont à respecter par le médecin investigateur qui mène la recherche. Il s’engage à respecter la législation en vigueur c’est-à-dire à conduire l’essai clinique selon les bonnes pratiques cliniques (BPC). Ces BPC établissent les lignes directives dans la gestion des essais cliniques (méthodologie précise, confidentialité des données des participants…). Elles définissent notamment les responsabilités du promoteur et de l’investigateur et garantissent aux participants (patients) leur protection, leur droit et leur sécurité.
Si l’on vous propose de participer à un essai clinique, le médecin vous présentera l’essai clinique (objectifs, procédures, risques…) et vous remettre une notice d’information écrite.
Cette notice décrit précisément l’objectif de l’essai clinique, les procédures à suivre (calendrier des visites à l’hôpital, prise de sang, radiologie, biopsie, hospitalisation, consultation avec le médecin…), les bénéfices potentiels et les risques éventuels, ainsi que vos droits et notamment celui de retirer votre accord de participation à tout moment. Vous aurez (et devez demander !) le temps suffisant pour lire cette notice, en parler si vous le souhaitez avec votre médecin traitant et vos proches. Vous pourrez ensuite lui poser toutes les questions que vous souhaitez avant de donner votre consentement de participation. Ce consentement est matérialisé par une signature conjointe d’un formulaire de consentement.
En droit de la santé ce consentement est dit libre et éclairé c’est-à-dire que le médecin est tenu de vous présenter clairement l’essai clinique et que vous acceptez sans contrainte d’y participer. Il n’y a pas de rémunération du patient pour sa participation à un essai clinique en cancérologie.
Essais cliniques : le témoignage de Nicolas
Après avoir appris brutalement qu'il avait un cancer, Nicolas décide au fil de son parcours de soin d'accepter de rentrer dans des essais cliniques au Centre Léon Bérard. Il témoigne aujourd'hui pour parler de leur efficacité et de leur importance pour la recherche.

Comment connaître les essais cliniques et la liste des essais cliniques ?
Pour en apprendre plus sur le principe des essais cliniques en particulier en cancérologie et vous informer sur la recherche, vous pouvez consulter les sites institutionnels suivants :
Les essais cliniques en cours au Centre Léon Bérard sont consultables ICI. N’hésitez pas à en discuter avec votre médecin référent.
Le site d’Unicancer : http://www.unicancer.fr/patients/la-recherche-clinique: https://notre-recherche-clinique.fr/
Tous les essais cliniques en cours au niveau international sont répertoriés sur le site (en anglais) clinicaltrial.gov
ZOOM SUR : le projet Trial Match 2 soutenu par le programme de coopération territoriale européenne Interreg France-Suisse 2021-2027
En rendant interopérables les données cliniques et les essais en cancérologie, le projet Trial Match 2 a pour but de permettre aux centres hospitaliers, universitaires et régionaux du bassin transfrontalier (Canton de Genève, Ain et Haute-Savoie) de favoriser un accès équitable et facilité aux essais cliniques les plus pertinents pour les patients suivis au Centre Léon Bérard ou aux Hôpitaux universitaires de Genève.
Pour ce faire, les partenaires utiliseront et adapteront un algorithme d’intelligence artificielle pour les rendre exploitables pour les hôpitaux. Ces outils s’appuieront sur les informations des dossiers des patients du territoire transfrontalier afin de proposer à ces derniers une liste d’essais cliniques locaux et adaptés. La solution sera totalement automatisée et pourra donc s’appliquer à n’importe quel volume de données au sein d’un hôpital. Les résultats du projet seront utilisables dans d’autres contextes de collaboration médicale et ouvriront la voie au rattachement d’autres hôpitaux.
Le projet ambitionne d’optimiser l’interopérabilité des données cliniques en oncologie en s’appuyant sur des solutions technologiques innovantes. Il vise aussi à renforcer l’attractivité de la région pour les industriels du médicament ainsi que pour les entreprises actives dans la recherche en oncologie. Par ailleurs, les partenaires industriels mettent en commun leurs compétences pour le développement, l’adaptation et l’exploitation de solutions d’analyse de données et de prédiction pionnières dans le domaine de l’oncologie. Ce projet est à l’initiative du clinicien-chercheur Loïc Verlingue, spécialisé dans les Essais cliniques de phase précoce (département d’Oncologie médicale).
Le Centre Léon Bérard et les Hôpitaux Universitaires de Genève sont chefs de file et les sociétés Coexya et Qiminfo sont partenaires.

